
Student News

October 4, 2021
Book your seat on zoom workshop.
Monday 11th October 7.30pm, Challenging Behaviour Specialist John Curran will be joining us.
December 16, 2020
€3K Access to Angelman Grant awarded.
We are delighted to have been awarded €3K today December 16th from the RTE Comic relief grant fund in conjunction with Community Foundation. As part of our Access to Angelman project we will be working to host virtual meeting and meetups in the coming months. More details to follow.
September 30, 2020
Entitlements for families
Booklet for families explaining entitlements for families. Kindly reproduced here with permission from Financialwellbeing.ie
September 20, 2019
2019 European Alliance Grants awarded
European Angelman Alliance Announce 2019 Research Grant winners
ASA proudly announced the 3 recipients: Dr. Ype Elgersma (Netherlands), Dr. Ugo Mayor (Spain) and Dr. Ben Philpot (USA) –
Dr. Ype Elgersma, Professor of Molecular Nuroscience, Erasmus Centre, Rotterdam, postulates that UBE3A plays a major role in the regulation of gene expression and sets out to identify respective genes.
Dr. Ugo Mayor,, Ikerbasque Research Professor, Bilbao has a daring and novel approach to identify and validate the substrates and cofactors (including DUB enzymes) of UBE3A and to test the viability of targeting DUB enzymes as a therapeutic strategy for AS. That intrigued both scientific board as well as the international alliance of parent organizations (Angelman Syndrome Alliance – ASA).
Dr. Ben Philpot,, Associate Professor, University of North Carolina, finetunes his last discovery by looking at quantifying sleep spindles from overnight EEGs as an Angelman syndrome biomarker.
Speaking on behalf of the scientific advisory board, Dr. Harald Sitte, : “The result of the 2018 call for research proposals has yielded a good combination of different research approaches which we hope to lead research on Angelman syndrome into a bright future. We congratulate the three awardees and look forward to follow their scientific work.”
The previous winners of the ASA Grants in 2014 and 2016 have been Dr. Geeske van Woerden, Dr. Ben Distel, Dr. Silvia Russo and Dr. Ben Philpot.
ASA is a global initiative of Angelman parent organizations.
Every country can have one representing AS parent-organisation participating in ASA. Our Scientific Advisory Board lends support and guidance to the members of the ASA parent organizations. Contact us if you wish to join or if you are interested to learn more about Angelman syndrome, ASA or the scientific challenges: www.angelman-alliance.org
For further information – www.angelman-alliance.org Communication: Betty Willemsen (betty.willemsen@ninafoundation.eu)
Scientific Board: Harald Sitte (harald.sitte@meduniwien.ac.at)
Angelman Ireland – admin@angelman.ie
June 25, 2019
Angelman Syndrome Alliance research published
- null
- This study was funded in part by ASA through research grants to Ben Distel, Geeske van Woerden and Ype Elgersma.
Location matters: A recent study provides a new viewpoint on how loss of UBE3A causes Angelman syndrome
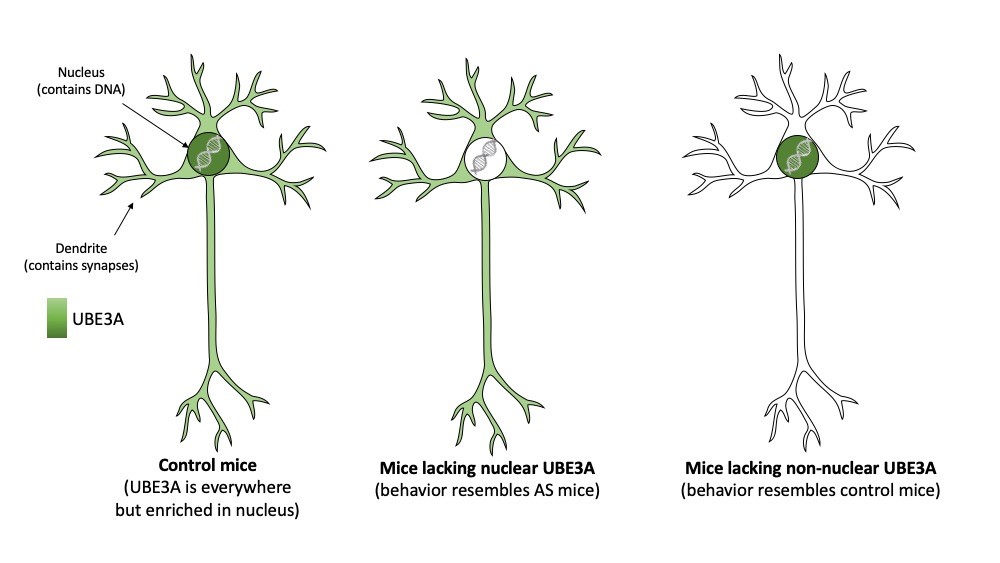
Angelman syndrome (AS) is caused by mutations or deletions that result in the loss of functional UBE3A protein. However, despite many years of research, it remains unclear where in the cell UBE3A performs its function and how the loss of UBE3A causes AS. A collaboration of three research groups in The Netherlands (Ben Distel, Amsterdam UMC, Amsterdam), Steven Kushner and Ype Elgersma (both at Erasmus MC, Rotterdam) has now resulted in a remarkable new viewpoint: UBE3A has a critical role in the nucleus and loss of nuclear UBE3A causes Angelman Syndrome. The study is published in this month’s issue of Nature Neuroscience.
Neurons form the computing core of our brain and communicate with each other through synaptic connections. Previous work of several research groups has established that the synapses of AS mouse models function suboptimal, and this deficit is likely underlying the severe neurodevelopmental delay. Since UBE3A is present in synapses, almost all research on UBE3A has focused on its role in the synapse, but its precise role remains elusive. However, UBE3A is also present in other places of the neuron, in particular it is highly abundant in the nucleus of neurons. The nucleus contains the DNA of the cell, and hence dictates the function of a cell. In the new study, the Dutch research team revealed precisely how UBE3A is able to get into the nucleus. They showed that UBE3A is able to bind to a protein called PSMD4, and that this binding is critical for bringing UBE3A into the nucleus.
Since nuclear localization of UBE3A is highly controlled, the team reasoned that the nuclear localization might be important for AS pathophysiology. Hence, they investigated if UBE3A mutations found in AS patients disrupt the targeting of UBE3A to the nucleus. Indeed, they describe three AS-associated UBE3A mutations in which UBE3A is no longer present in the nucleus.
To investigate this further, the team made use of the observation that the UBE3A protein comes in two different sizes (called isoforms). The authors showed that only the shorter form is present in the nucleus. The longer form of UBE3A is absent in nuclei and dispersed throughout the neuron (for instance in synapses). To further investigate the importance of the two UBE3A isoforms and their differential localization, the researchers generated two mouse models that either made the nuclear (short) UBE3A protein or the non-nuclear (long) UBE3A protein. Consistent with an important role of UBE3A in the nucleus the researches showed that mice which specifically lack nuclear UBE3A, highly resembled AS mice that lack UBE3A altogether. Not only did these mice show behavioral deficits, also the synapses of these mice were no longer functioning properly. In contrast, mice that lacked the non-nuclear (long) UBE3A appeared unaffected.
The authors indicate that these findings change the current view of how UBE3A causes AS. The team suggests that future studies should elucidate the precise role of UBE3A in the nucleus and how this role relates to the pathophysiology of AS. This knowledge is important to develop new treatments.
This study was funded in part by ASA through research grants to Ben Distel, Geeske van Woerden and Ype Elgersma.
Full text available at:
Avagliano-Trezza et al., Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome, Nature Neuroscience, doi: 10.1038/s41593-019-0425-0.
April 15, 2019
Mick Falvin to perform at fundrasier – Naomh Malachi GFC club Saturday 15th June.
The family of Shane Gorman are hosting an evening of music with the fantastic Mick Flavin – 16th June. Tickets $10 with all proceeds kindly being donated to ASI. For details and if you can support or donate prizes contact Seamus – 086 325 6906 or email ASI at admin@angelman.ie
February 13, 2019
IAD 2019
Dublin family meet up details : Sunday 17th Marley park playground 1pm.
Belfast ASI/ASSERT Sunday 17th Kids Together, Stewartstown Road Belfast 12.30-3.30.
For more details email admin@angelman.ie

